
O que é a Doença de Pompe?
A doença de Pompe é uma doença lisossomal de sobrecarga causada pela atividade insuficiente do ácido á-glucosidase.1 Esta enzima lisossómica é responsável pela degradação do glicogénio intralisossómico, o qual representa apenas uma pequena percentagem (1-3%) do glicogénio celular total.
A deficiência enzimática resulta numa acumulação de glicogénio lisossomal em múltiplas células e tecidos. Eventualmente, esta sobrecarga conduz a lesões e disfuncionalidades a nível celular, particularmente nos tecidos cardíaco, respiratório, esquelético e muscular.1,2
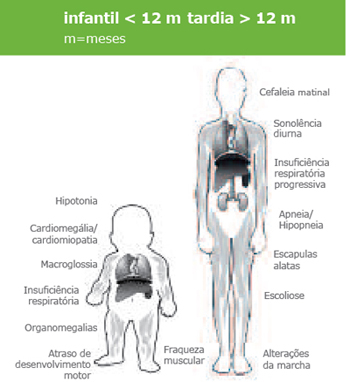
As manifestações clínicas da doença de Pompe são extremamente variáveis. No extremo mais grave do espectro desta doença, a insuficiência cardio-respiratória conduz à morte, geralmente durante o primeiro ano de vida, em 80% das crianças (que tipicamente apresentam envolvimento cardíaco e músculo–esquelético).
Nos casos em que a doença se manifesta numa fase mais tardia, a fraqueza muscular esquelética e respiratória progridem inexoravelmente e podem conduzir a uma dependência de cadeira de rodas e/ou de ventilador, e num momento final, à morte, que ocorre entre o início da infância e o final da idade adulta.
A doença de Pompe também tem sido referida como uma doença de sobrecarga de glicogénio do tipo II, glicogenose tipo II e deficiência em ácido maltase, em virtude de se tratar da forma mais grave das 12 doenças de sobrecarga de glicogénio que se conhecem.
A doença de Pompe é uma alteração autossómica recessiva com uma penetrância variável. As estimativas actuais colocam a incidência total desta doença na ordem de 1 em cada 40.000 nados vivos.3,4 Pode-se extrapolar que a prevalência mundial desta doença pode estar algures entre os 5.000 e os 10.000 casos.
Manifestações
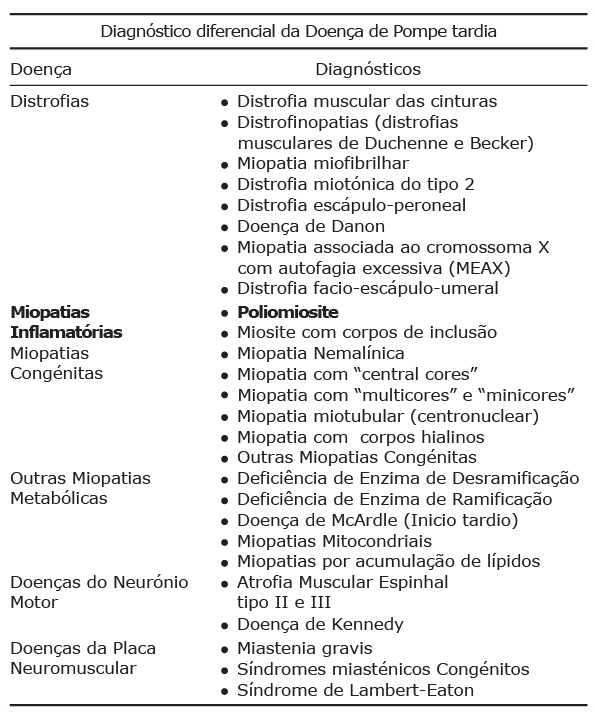
Diagnóstico diferencial da doença de Pompe
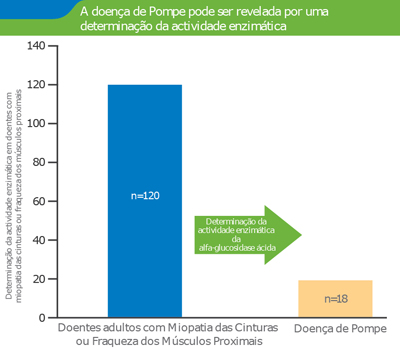
Incidência de doença de Pompe
Incidência elevada de Doença de Pompe em doentes com miopatia das cinturas ou fraqueza dos músculos proximais não explicadas. 1
- Em 15% dos doentes adultos com miopatia das cinturas ou fraqueza dos músculos proximais suspeitos de sofrer de doença de Pompe foi obtida confirmação de diagnóstico. 1
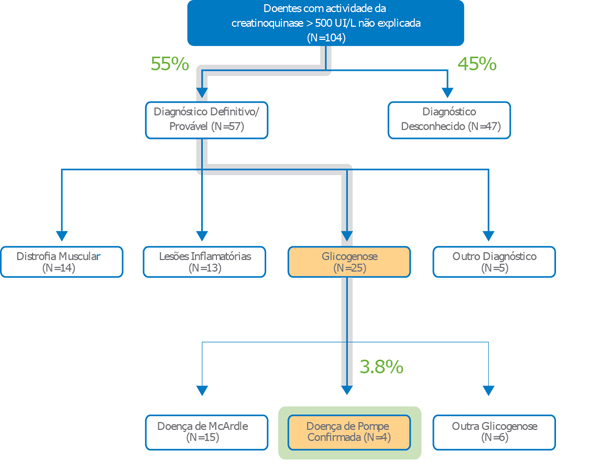
Incidência de doença de Pompe em doentes com níveis elevados não explicados de CK2
- Em 3,8% dos doentes com níveis elevados de e não explicados de CK foi confirmado o diagnóstico de doença de Pompe.2
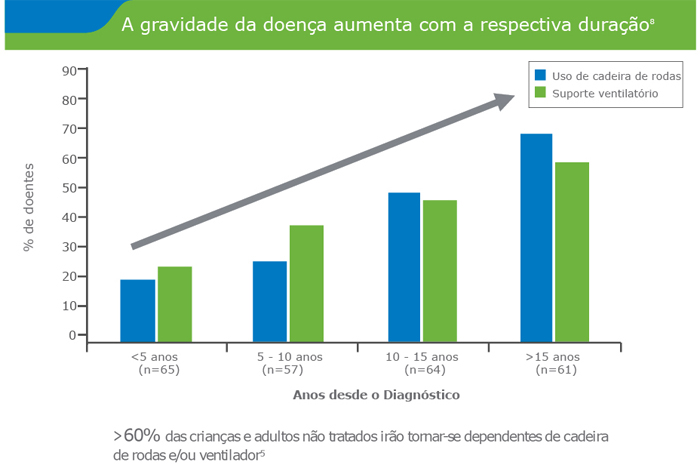
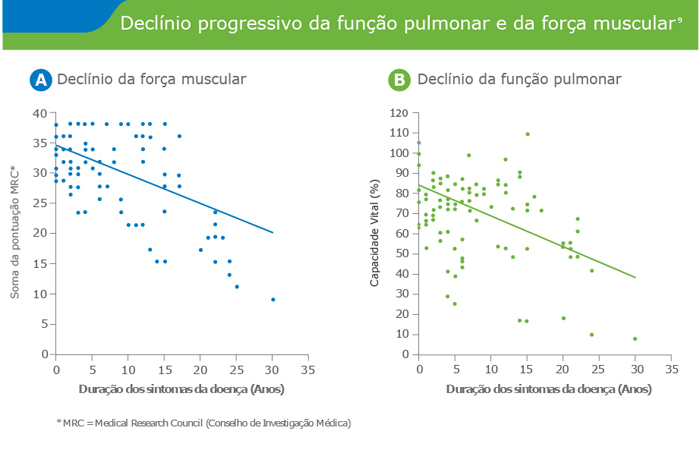
- A doença de Pompe é uma patologia Neuromuscular progressiva, debilitante e frequentemente fatal
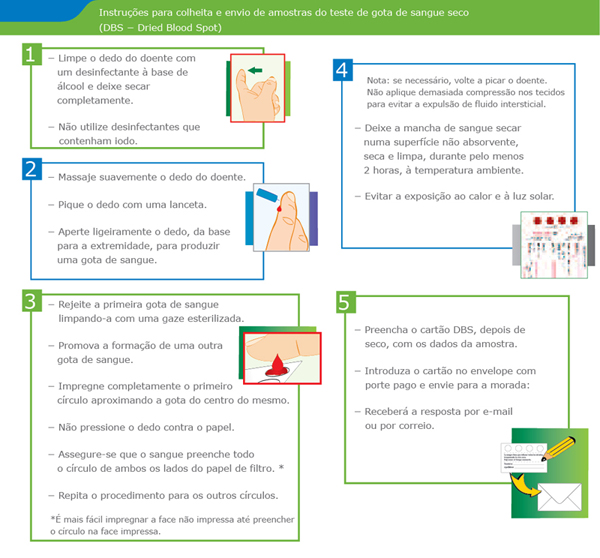
O teste de Diagnóstico é simples
- Uma revisão de biópsias musculares em doentes adultos com diagnóstico confirmado de doença de Pompe revelou que apenas 10-50% das fibras apresentavam vacuolização. 10
- A AAMNE recomenda a determinação da deficiência parcial ou total da atividade enzimática da alfa-glucosidase ácida (GAA) como padrão de eleição para o disgnóstico.9
- Uma análise rápida e simples (linfócitos ou leucócitos numa gota de sangue seco), permite um disgnóstico precoce e preciso da doença de Pompe.
Referência Bibliográficas
- Hirschhorn R, Reuser A. Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR BA, Sly WS, Valle D (eds). The metabolic and molecular bases of inherited disease, 8th Ed. New York, New York: McGraw-Hill 2001:3389-420.
- Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144(5):35-43.
- Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998;79(1):69-72.
- Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999;7(6):713-6.
- American Association of Neuromuscular & Electrodiagnostic Medicine. Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009 Jul;40(1):149-60.
- Goldstein JL, Young SP, Changela M, Dickerson GH, Zhang H, Dai J, Peterson D, Millington DS, Kishnani PS, Bali DS. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve. 2009 Jul;40(1):32-6.
- Fernandez C, de Paula AM, Figarella-Branger D, Krahn M, Giorgi R, Chabrol B, Monfort MF, Pouget J, Pellissier JF. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology. 2006 May 23;66(10):1585-7.
- Hagemans ML,Winkel LP, HopWC, Reuser AJ, Van Doorn PA, Van der Ploeg AT. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology. 2005 Jun 28;64(12):2139-41.
- Van der Beek NA, Hagemans ML, Reuser AJ, HopWC, Van der Ploeg AT, Van Doorn PA,Wokke JH. Rate of disease progression dur¬ing long-term follow-up of patients with late-onset Pompe disease. Neuromuscul Disord. 2009 Feb;19(2):113-7. Epub 2008 Dec 11.
- Schoser BG, Müller-Höcker J, Horvath R, Gempel K, Pongratz D, Lochmüller H, Müller-FelberW. Adult-onset glycogen storage disease type 2: clinico-pathological phenotype revisited. Neuropathol Appl Neurobiol. 2007 Oct;33(5):544-59. Epub 2007 Jun 15.
Para mais informações depmedico-pt@genzyme.com